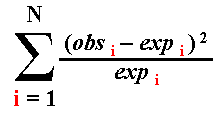
| d.f. | p=0.9 | p=0.5 | p=0.1 | p=0.05 | p=0.01 |
| 1 | 0.016 | 0.455 | 2.706 | 3.841 | 6.635 |
| 2 | 0.211 | 1.386 | 4.605 | 5.991 | 9.210 |
| 3 | 0.584 | 2.366 | 6.251 | 7.815 | 11.345 |
| k (# heads) | p(k|pN) |
| 0 | 0.00000095 |
| 1 | 0.00001907 |
| 2 | 0.00018120 |
| 3 | 0.00108719 |
| 4 | 0.00462055 |
| 5 | 0.01478577 |
| 6 | 0.03696442 |
| 7 | 0.07392883 |
| 8 | 0.12013435 |
| 9 | 0.16017914 |
| 10 | 0.17619705 |
| 11 | 0.16017914 |
| 12 | 0.12013435 |
| 13 | 0.07392883 |
| 14 | 0.03696442 |
| 15 | 0.01478577 |
| 16 | 0.00462055 |
| 17 | 0.00108719 |
| 18 | 0.00018120 |
| 19 | 0.00001907 |
| 20 | 0.00000095 |
| k (# heads) | p(k|pN) |
| 0 | 0.00000095 |
| 1 | 0.00001907 |
| 2 | 0.00018120 |
| 3 | 0.00108719 |
| 4 | 0.00462055 |
| 5 | 0.01478577 |
| 6 | 0.03696442 |
| 7 | 0.07392883 |
| 8 | 0.12013435 |
| 9 | 0.16017914 |
| 10 | 0.17619705 |
| 11 | 0.16017914 |
| 12 | 0.12013435 |
| 13 | 0.07392883 |
| 14 | 0.03696442 |
| 15 | 0.01478577 |
| 16 | 0.00462055 |
| 17 | 0.00108719 |
| 18 | 0.00018120 |
| 19 | 0.00001907 |
| 20 | 0.00000095 |
| Outcome Class | Observed Number of Occurrences of Outcome | Probability of Outcome Class | Expected Number of Occurrences of Outcome | (obs-exp)2 / exp |
| "heads" | 12 | 0.5 | 0.5 x 20 = 10.0 | (12-10)2 / 10 = 0.4. |
| "tails" | 8 | 0.5 | 0.5 x 20 = 10.0 | (8-10)2 / 10 = 0.4. |
| Sum | 20 | . | 20.0 | 0.8 |
| Outcome Class | Observed Number of Occurrences of Outcome | Probability of Outcome Class | Expected Number of Occurrences of Outcome | (obs-exp)2 / exp |
| "heads" | 4,865 | 0.5 | 0.5 x 10,000 = 5,000.0 | (4,865-5,000)2 / 5,000 = 3.645 |
| "tails" | 5,135 | 0.5 | 0.5 x 10,000 = 5,000.0 | (5,135-5,000)2 / 5,000 = 3.645 |
| Sum | 10,000 | . | 10,000.0 | 7.290 |
| Phenotype | Observed | Expected | Obs - Exp | (Obs - Exp)2 Exp |
| AB | 25 | 64.00 | -39.00 | 23.77 |
| Ab | 25 | 16.00 | 9.00 | 5.06 |
| aB | 25 | 16.00 | 9.00 | 5.06 |
| ab | 25 | 4.00 | 21.00 | 110.25 |
| A gene phenotype | |||||
| A | a | ||||
| B gene pheno- type | B |
22 AB offspring |
18 aB offspring | 22 + 18 = 40 B offspring | Frequency of B allele 40/100 = 0.40 |
| b |
27 Ab offspring |
33 ab offspring | 27 + 33 = 60 b offspring | Frequency of b allele 60/100 = 0.60 | |
| 22 + 27 = 49 A offspring | 18 + 33 = 51 a offspring | 100 Total offspring | |||
| Frequency of A allele = 49/100 = 0.49 | Frequency of a allele = 51/100 = 0.51 | ||||
| Phenotype | Observed | Expected | Obs - Exp | (Obs - Exp)2 Exp |
| AB | 22 | 19.60 | 2.40 | 0.29 |
| Ab | 27 | 29.40 | -2.40 | 0.20 |
| aB | 18 | 20.40 | -2.40 | 0.28 |
| ab | 33 | 30.60 | 2.40 | 0.19 |
| Phenotype | Observed | Expected | Obs - Exp | (Obs - Exp)2 Exp |
| Red | 200 | 160 | 40 | 10.00 |
| White | 400 | 360 | 40 | 4.44 |
| Pink | 400 | 480 | -80 | 13.33 |